Protein kinases constitute a vast and diverse family of enzymes that play a significant role in mediating cellular activities by regulating energy transfer between adenosine triphosphate (ATP) and other substrates. The structural elements that are shared across kinases include an amino acid (DFG) motif containing an activation loop that regulates kinase activity. Many diseases, including cancer, can be attributed to an impairment in kinase activity. This, in turn, adversely affects downstream signalling pathways. As a result, kinase-targeted therapy presents a hot area for research, with several small molecule kinase inhibitors underway or in the market.
Type‑I kinase inhibitors are known as ATP-competitive inhibitors — they compete with ATP in binding to a gatekeeper amino acid residue in what is known as the hinge region of the kinases. However, the occurrence of mutations at this site, commonly threonine to methionine substitutions, renders this class of inhibitors ineffective. Consequently, type-II inhibitors, which can bypass the kinase gatekeeper mutations, have been developed. The mechanism by which these inhibitors bind to the kinases despite mutations remained ambiguous until now. Jagannath Mondal and co-workers, from Tata Institute of Fundamental Research, Hyderabad (TIFR‑H) and Columbia University, USA performed atomistic molecular dynamics simulation studies to understand the binding mechanisms of these mutant-resistant kinase inhibitors. The results of their studies have been published in the Journal of the American Chemical Society.
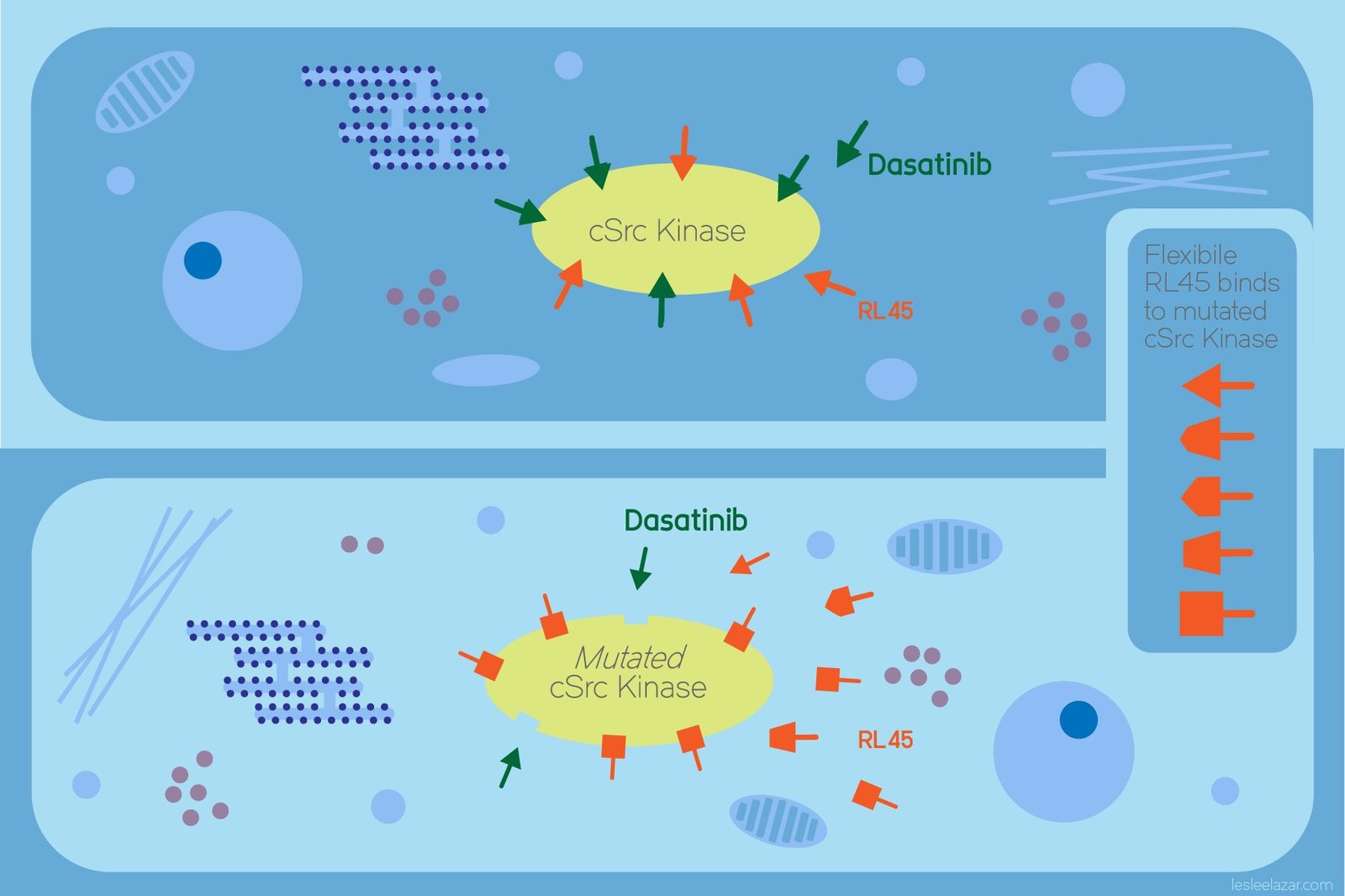
The researchers compared the behaviour of a type I inhibitor called Dasatinib and a type II inhibitor called RL45 toward cSrc-kinase, with and without a gatekeeper residue mutation. cSrc-kinase is a cytoplasmic kinase that regulates several cell signalling activities, whose aberrant activity has been strongly implicated in cancer. In this study, the researchers used a state-of-the-art computational technique called “free energy perturbation with replica exchange solute tempering (FEP/ REST) simulations”. This enhanced sampling technique was employed to compute protein-ligand binding affinities, a measure of the strength of attraction between a receptor and ligand, for both inhibitors.
The results of the powerful FEP/REST simulations correlated with previous experimental findings, showing that the gatekeeper residue mutation caused an unfavourable change in binding free energy between Dasatinib and the kinase. RL45, the type-II inhibitor, on the other hand, could bind effectively to both the wild and mutated gatekeeper residue. Further analysis revealed that differences in ligand flexibility influenced the binding of both inhibitors to the mutant kinase. The failure of the type‑I inhibitor, Dasatinib, stemmed from its lack of flexibility — inability to avoid steric clashes and loss of a crucial ligand−pocket hydrogen bond upon the T338M mutation. A flexible central phenyl ring in RL45, however, overcame these problems and was able to bind to the mutant.
“Our results point towards the development of kinase inhibitors which are not conformationally rigid and can reorient themselves in the wake of gatekeeper residue mutations,” stated the researchers, in an email correspondence. Their study establishes that FEP/REST could be used as an efficient simulation technique to perform high throughput screening of potential drug candidates against these mutations.
The scope for future studies can be divided into two steps. “First, we would like to study whether DFG-in vs DFG-out conformation of kinase has any allosteric roles to play in withstanding gatekeeper residue mutation,” said the investigators. Furthermore, they aim to analyse the efficacy of the inhibitor by evaluating its residence time in the native binding pose. “We are on course to investigate the unbinding mechanisms and rate-determining steps of the kinase inhibitor from its bound pose.” This would provide a clearer picture for designing better kinase inhibitors, hence greatly contributing to targeted kinase therapy for various diseases.